

The Great Convergence: FDA Transitions from QSR to QMSR
For decades, the global medical device industry has operated on regulatory islands, forcing manufacturers to manage a fractured compliance landscape. A producer in India historically had to maintain three distinct Quality Management Systems (QMS) to access the world’s largest markets: the US FDA’s 21 CFR Part 820 (QSR), the European Union’s CE-MDR/IVDR, and domestic standards governed by the CDSCO and ICMR. As of February 2, 2026, this fragmented era has ended. The FDA’s final transition to the Quality Management System Regulation (QMSR) formally incorporates ISO 13485:2016 by reference, establishing a singular technical language for a global med-tech market now valued at over $600 billion.
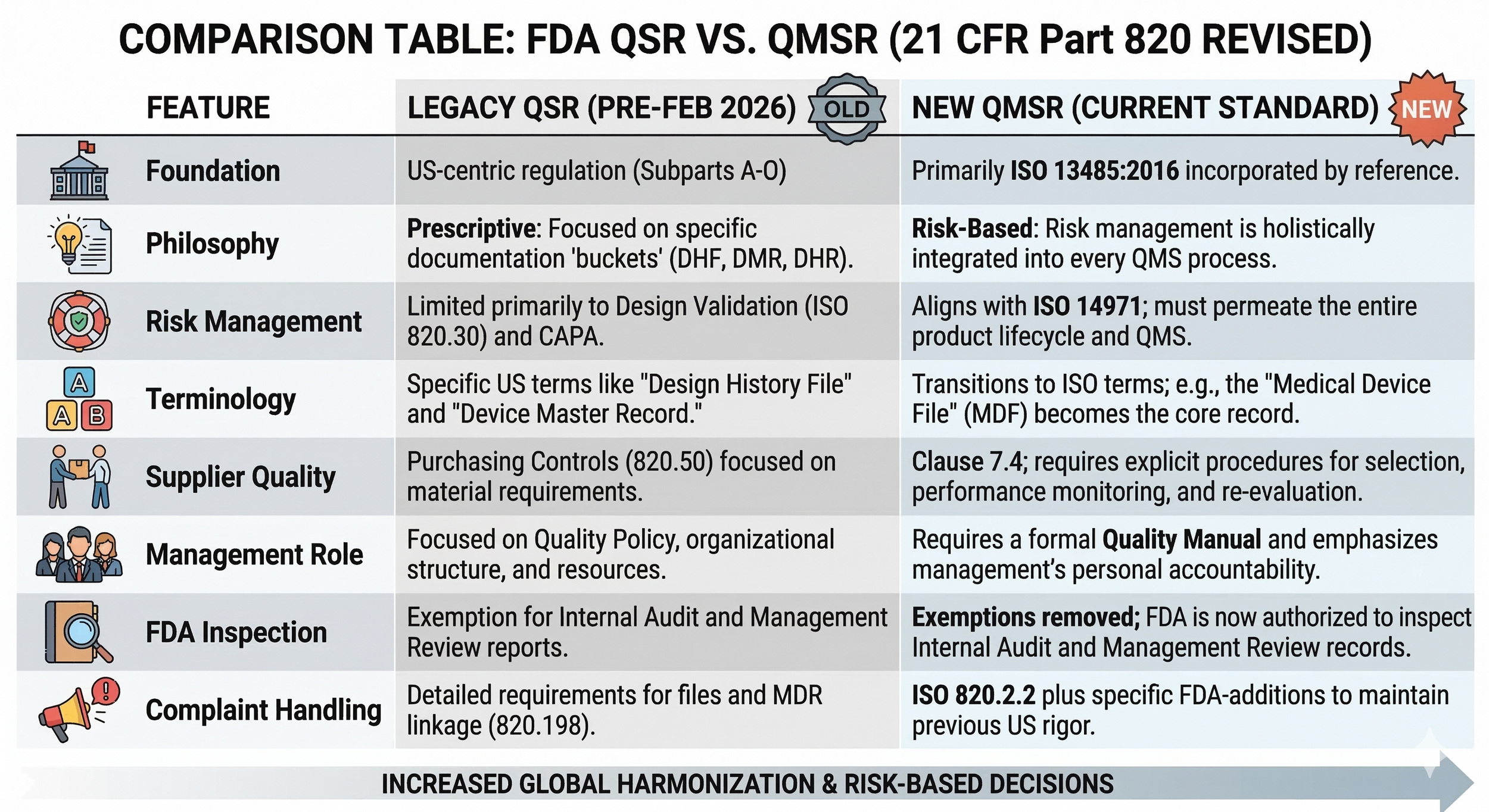
The most profound shift is philosophical. The legacy QSR was prescriptive, dictating exactly what to document, in contrast, the QMSR is risk-based, mandating that ISO 14971 (the global standard for risk management) permeates every operational layer. While the old QSR limited formal risk analysis primarily to the design phase, the QMSR requires a "risk rationale" for every process - from supplier audits to employee training. This alignment with European CE standards, which is also harmonised with the ISO 13485 system, eliminates a "Compliance Tax" that previously added an estimated 10–15% to the overheads of cross-border medical trade.
For the Indian MedTech ecosystem (currently the 4th largest in Asia) this is a strategic windfall. The CDSCO has aggressively aligned its Medical Device Rules (MDR 2017) with these international benchmarks. Consequently, when the ICMR validates a high-sensitivity RDT, it does so within a framework that mirrors global expectations for analytical sensitivity and clinical specificity. This harmonization creates regulatory fungibility for global inventory; since the underlying technical dossiers are now standardized. This means, a batch of lot-verified monitors can be redirected from an ASEAN port to an African hub without the traditional six-month delay for redundant compliance re-documentation.
The consequences for emerging markets are systemic, with the FDA and EU now converging on ISO 13485, regional regulators can implement reliance pathways to accelerate approval cycles for devices already vetted under this unified global audit, a radical shift from historical 18-month lead times. Access to high-quality, reliable medical devices and equipment will now be easier all across the world and for manufacturers, operating in different areas of the globe becomes a simpler endeavour.